以前のコラムで紹介した通り、再生医療等製品の臨床開発、臨床応用に関しては、薬機法・安確法の整備がなされてきました。
> 再生医療に関する法律 その1(別ウインドウで開きます)
> 再生医療に関する法律 その2(別ウインドウで開きます)
一方で、再生医療に使用する細胞加工物、特にヒト幹細胞由来細胞製品の品質・安全性評価については、製品の種類・特性、また臨床における適用法などを踏まえた評価が必要であり、その試験内容はケース・バイ・ケースでした。
そのような中、2020年に日本医療研究開発機構(AMED)の再生医療実用化基盤整備促進事業の1課題成果として「ヒト幹細胞等加工再生医療製品の品質及び安全性等評価に共通の基本となる技術要件・基準・留意事項(ミニマム・コンセンサス・パッケージ)」が提言され、多くの製品の開発に共通する技術要件や基準が示されました。
今回は、その中でもヒト幹細胞由来製品の造腫瘍性試験に関する技術要件・基準等ついてご紹介したいと思います。
低分子医薬品や生物学的製剤では、投与した薬剤が患者の細胞に作用しがんを引き起こす「がん原性」というリスクがあります。
この「がん原性」とは異なり、ヒト細胞製品(特にヒト多分化能性細胞由来製品)では投与した細胞製品自体が異常増殖して腫瘍を形成するというリスクがあります。この腫瘍形成リスクの原因の1つとして、製造過程(主に分化過程)における細胞の形質転換(足場非依存的増殖能獲得)というものが挙げられます。
また、ES/iPS細胞などの多能性幹細胞は、自己複製能力(細胞分裂して、自分と同じ性能を持つ未分化なES/iPS細胞を複製する能力)を持っているため、細胞製品中に未分化ES/iPS細胞が残存していると、細胞製品投与後にこれらの未分化細胞が自己複製(増殖)してテラトーマ(奇形腫)を形成するリスクがあります。
再生医療等製品の品質・安全性の評価では、これらの腫瘍形成リスクを評価するために、in vitro/in vivo試験が求められることになります。
例えば、多能性幹細胞(ES/iPS細胞)を神経細胞に分化させた製品の場合、最終製品に「①形質転換細胞(製造(分化)過程で遺伝子に傷が入り、がん化した細胞)」、及び「②神経細胞に分化せず未分化のまま残ったES/iPS細胞」が残存していると、投与後に増殖して腫瘍を形成するリスクがあります(図)。
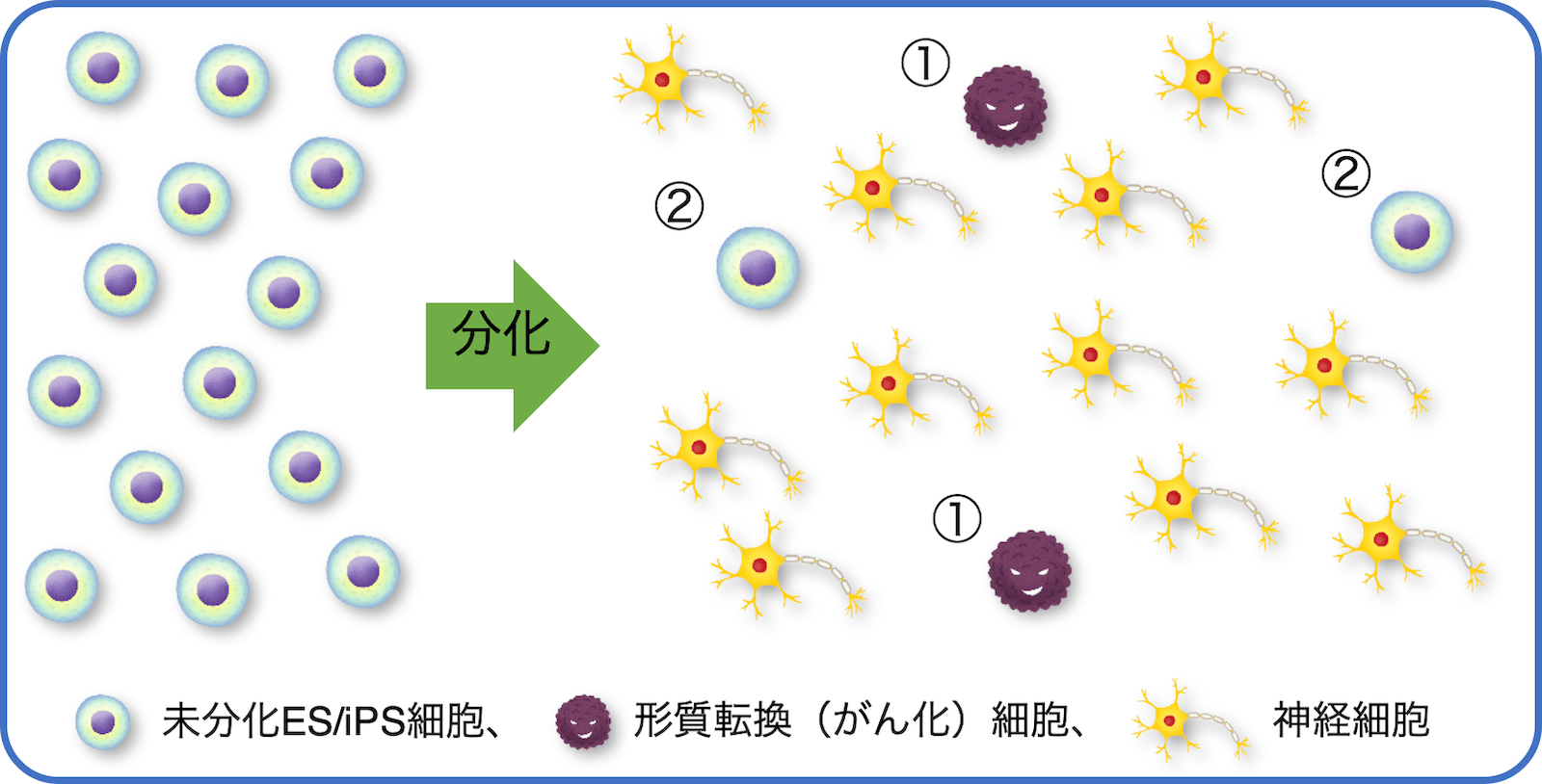
多能性幹細胞由来製品における未分化残存細胞の評価は主にin vitro試験(PCRや長期培養法)により未分化残存細胞(未分化マーカー)の検出を行い、そのリスク評価を行います。
形質転換細胞の評価は軟寒天コロニー試験(足場非依存的増殖能の確認)または生着部位(臨床での移植部位)における腫瘍形成能評価を含めたin vivo試験を行います。
In vivo試験の実施に当たっては、①使用動物、②使用製品、③投与経路、④観察期間が試験計画を立てる上で重要となります。
①使用動物としては、長期の生着が見込める免疫不全動物(NOG/NSGマウスなど)の使用が提言されています。大動物は免疫不全動物の供給が今のところ不安定であり、試験には免疫抑制剤の投与が必要なこと、長期の観察には向かないことから不向きとされています。
②使用製品としては、最終製品(同等品)を用いることとされており、③投与経路については臨床適用投与経路を用いることが提言されています。
また、④観察期間は細胞の残存期間等を考慮した期間設定となりますが、最長、動物が死亡するまで(寿命と判断できるまで)の設定となります。
特に、in vivo試験は長期に渡ることが多いため、開発計画に大きなインパクトを与えることから、事前の綿密な試験計画等が必要となります。
レメディグループでは基礎研究・前臨床開発段階における開発計画・当局相談のサポートも実施しており、これまでにも各種PMDA相談(再生医療等製品戦略相談、再生医療等製品等の品質及び安全性に係る相談、カルタヘナ法関連相談など)のサポートの実績がございます。
> レメディグループの再生医療について(別ウインドウで開きます)
このように、レメディグループでは、再生医療等製品開発の基礎研究・前臨床の段階から臨床の段階まで対応可能な専門チームを編成しており、一貫したサポート対応が可能です。
再生医療等製品の開発に関するご相談がございましたら、レメディグループまでお問い合わせください。