The Act on Ensuring the Safety of Regenerative Medicine, etc. (hereinafter referred to as the "Act on Ensuring the Safety of Regenerative Medicine, etc.") enacted in 2013 has placed a net of pre-regulation over free medical treatment using cell processing products, which was previously unregulated as part of the scope of doctors' discretionary authority.
The Act for Securing the Safety of Regenerative Medicine also regulates regenerative medicine conducted as clinical research.
However, in order to avoid the “doubling” of regulations, treatments already regulated under other laws are exempted.
Specifically, clinical trials for processed cells expected to be regenerative medicine products and regulated by the Pharmaceutical and Medical Device Act, and treatments regulated by the “Act on the Promotion of Proper Provision of Hematopoietic Stem Cells” used in the transplantation and its enforcement regulation, are exempted from the Act on the Safety of Regenerative Medicine.
Also, only specific cell-processed products except regenerative medicine products defined in the Pharmaceutical and Medical Device Act are regulated by the Act on the Safety of Regenerative Medicine. This regulation is not applicable to gene therapies such as plasmid and virus vectors. Clinical research of gene therapies are regulated by the Clinical Trials Act.
There is a concern because there are no laws regulating gene therapies in private practice (uninsured).
Chart 1 Applicable law
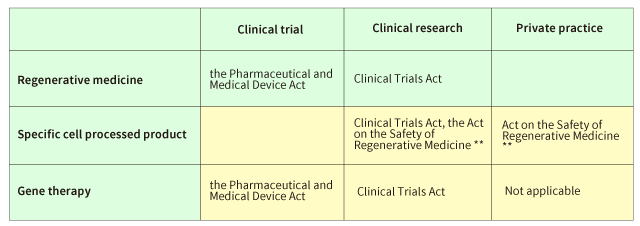
** Act on the Safety of Regenerative Medicine
Categories of Regenerative Medicine Technologies in the Act on the Safety of Regenerative Medicine
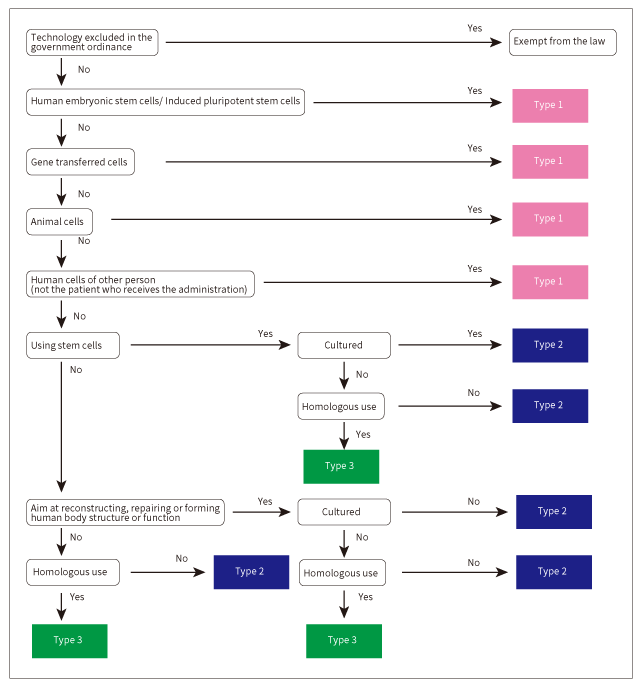
In the Act on the Safety of Regenerative Medicine, regenerative medicine technologies are categorized into Type 1, Type 2 and Type 3 depending on the risk.
Pluripotent stem cells including ES cells and iPS cells, gene transfer cells, regenerative medicine technologies using animal cells and allogeneic cells fall into the Type 1. In case of autologous stem cells, cultured cells or non-cultured cells for non-homologous use fall into the Type 2. Cells that are not cultured and for homologous use, fall into Type 3.
In the case of autologous cells, the following fall into Type 2:
- Cultured somatic cells with the aim of reconstruction, repair or formation of human body structure or function
- Non-cultured somatic cells for non-homologous use
- Non-cultured cells for homologous use fall into Type 3. Even in case of technologies in which somatic cells are used and is not aimed at reconstructing, repairing or forming human body structure or function, cells for non-homologous use fall into Type 2, and cells for homologous use fall into the Type 3.
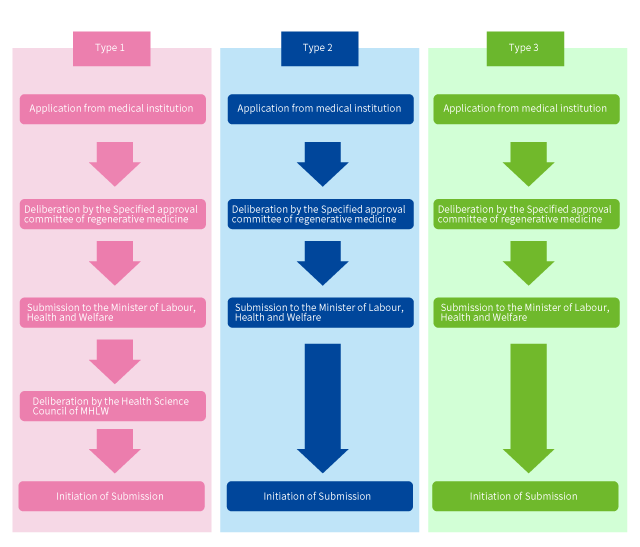
When a regenerative medicine treatment is planned in clinical research or private practice, it is necessary to seek the approval of the Committee of Regenerative Medicine which will deliberate and approve the treatment, and then submit an application to the Minister of Labour, Health and Welfare.
lso, the disclosure of information regarding the treatment is required. Type 1 and Type 2 are deliberated by the specified approval committee of regenerative medicine, and Type 3 is deliberated by the approval committee of regenerative medicine. Also, as for the Type 1, it is necessary to obtain the opinion of the Health Science Council of MHLW after the submission, and a 90-day provision limitation has been set for this period.
Disclaimer: The information provided is not intended to provide any pharmaceutical or medical device regulatory law advice; instead, all information and content are for general informational purposes only.